A good electronic records managment program will not just impress regulators. It also can boost profitability.
Pharmaceutical Technology-06-01-2005
This article describes the Part 11 requirements that companies must address when automating their quality management processes and offers practical tips for compliance.
Glossary of computer and software terms.

Nearly eight years have elapsed since the US Food and Drug Administration's 21 CFR Part 11 regulations on the use of electronic records and electronic signatures went into effect (1). In Sept. 2003, FDA issued a guidance document covering the scope and application of Part 11, which described how the agency intends to interpret and enforce the requirements during its ongoing re-examination of the regulations (2). Many in the pharmaceutical industry view the guidance as a positive development that will lead to a simplified FDA approach to Part 11 and a significant reduction in the industry's compliance burden. But this shift in FDA's interpretation and its intended use of enforcement discretion has not ended the controversy and confusion surrounding Part 11 and its requirements.
The confusion surrounding the application of 21 CFR Part 11 can be solved by focusing on the common rationale that both protects the public health and drives the fundamental basis of the industry's business: good science.

The use of quality management software (QMS) to automate manufacturing quality processes quickly is becoming an industry-wide initiative. Companies are turning to commercial off-the-shelf systems (COTS) to simplify implementation and validation efforts. Regardless of the system selected, the automation of these critical quality processes is subject to electronic records and electronic signatures (ER/ES) requirements, as set forth in the 21 CFR Part 11 regulation (1). Therefore, companies must adopt a comprehensive but manageable approach to Part 11 compliance as they begin to automate the processes that support product quality and efficacy to ensure they meet the requirements of good manufacturing practices and all predicate rules.
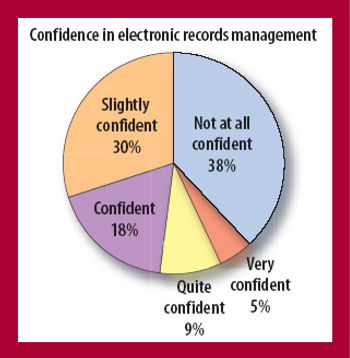
Pharmaceutical firms have every reason to feel confused and even a bit bewildered by what the US Food and Drug Administration expects from them when it comes to controlling electronic records. At times over the past nine years it has been a bit like riding a roller coaster that you weren't allowed to get off.
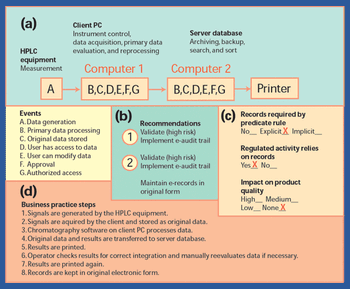
In 1997, the US Food and Drug Administration issued a regulation"Rule 21 CFR Part 11," that provides criteria for the acceptance of electronic records, electronic signatures, and handwritten signatures (1). FDA issued the guidance in response to requests from the industry. With this regulation, electronic records can be equivalent to paper records and handwritten signatures. The rule applies to all industry segments regulated by FDA that include good laboratory practice, good clinical practice, and current good manufacturing practice.

Paper notebooks are the accepted method for recording laboratory data and the ideas generated from that information in the pharmaceutical, biotech, and chemical industries. Nonetheless, the revolution in digital data processing has improved the way data is created, organized, and managed electronically, whether in the form of analytical data, images, documents, or multimedia files. The preservation of such information into a digital form offers the potential for online storage and retrieval, efficient search processes, and worldwide data transmission.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Novo Nordisk Sues Eli Lilly Over Zepbound and Mounjaro Ads, Alleging Omitted GLP-1 Dosing Data
2
What Jideytro's Approval Means for Drug Developers
3
Midyear 2026 Check in: Abzena’s Campbell Bunce on Trends in Antibody Drug Conjugates
4
Midyear 2026 Check-In: AI Investment Skipped Pharma's Costliest Operational Problem
5