Evaluating Impurities in Drugs (Part I of III)
In Part I of a three-part article, the authors discuss what constitutes an impurity and the potential sources of impurities in APIs and finished drug products.
To ensure the quality of APIs and finished drug products, impurities must be monitored carefully during process development, optimization, and process changeover. The isolation, characterization, and control of impurities in pharmaceutical substances are being reviewed with greater attention based on national regulatory and international guidelines. In Part I of this article, the authors examine the different types and sources of impurities with specific examples.



Definition and sources of impurities
An impure substance may be defined as a substance of interest mixed or impregnated with an extraneous or usually inferior substance. The greatest financial impact on the cost of a drug substance often is found in the final preparation process. Product yield, physical characteristics, and chemical purity are important considerations in the manufacture of the active ingredient, the formulation of the dosage form, and the manufacture of the finished drug product. Processes to control the preparation of the drug substance and drug product must be disclosed to FDA as part of a new drug application. If production batches do not meet the purity and impurity specifications required, the manufacturer must attempt to upgrade materials by rework procedures, which are costly because they consume drug substance and resources and prevent the preparation of other batches of drug substance. The sources and types of impurities can be illustrated by considering a general flow scheme for manufacturing drugs. The formation of impurities is interconnected with each stage as shown in Figure 1.



Figure 1: Schematic representation of impurity-formation pathways for APIs and finished drug products. DMF is drug master file.
In short, any material that can affect the purity of an API or finished drug product is considered an impurity. Impurities arise from various sources, which commonly include starting material(s), intermediates, penultimate intermediates, byproducts, transformation products, interaction products, related products, degradation products, and tautomers.
Starting material(s)
Impurity control in starting materials used to manufacture APIs has long been expected by regulatory agencies (1). An API starting material is a raw material, intermediate, or API that is used in the production of an API and that is incorporated as a significant structural element into the API. API starting materials normally have defined chemical properties and structure (2). An FDA draft guidance, Drug Substance: Chemistry and Manufacturing Controls Information, reflects the concern that starting materials should be selected and controlled such that any potential future changes to the quality of the starting material would have an insignificant impact on the safety, identity, purity, or quality of the drug substance (3). Based upon the principles outlined in this FDA draft guidance and ICH guidelines for process understanding and control over potential adverse effects on the quality of the produced drug substance, the following framework has been offered for the selection of starting materials:
- Appropriate, discriminating methodology is used to determine the quality of the starting material.
- Specifications are appropriate to ensure quality of the API.
- The impact of the starting material quality on API quality is understood and controlled.
- The starting material is available commercially and is incorporated into the new drug substance as an important structural element.
- The starting material is characterized, and stability is well understood.
- The starting material is a compound whose name, chemical structure, chemical and physical characteristics and properties, and impurity profile are well defined in the chemical literature (4).
Because of the starting materials' potential impact on the quality of an API, stricter requirements for a starting material arise based on the proximity in the API synthesis of the starting material to the final API. For example, fluoronitrobenzene is a key starting material for the API olanzapine. If the 2-4-difluoronitrobenzene impurity is present in the key starting material, the same will be converted under reported conditions to 8-fluoro-olanzapine, a nonpharmacopeial impurity (US Pharmacopeia [USP] method, relative retention time [rrt] 1.07). The 2,4-difluoronitrobenzene is carried forward along with the fluoronitrobenzene, resulting in analogous compounds up to the final stage.
In another example, N-[6-(4-phenylbutoxy)hexyl)] benzenemethanamine (see Figure 2) is a drug master file (DMF) starting material for the selective long-acting -2-adrenoreceptor agonist salmeterol. The drug is used clinically as an inhaled bronchodilator for treating asthma and chronic bronchitis (5, 6).



Figure 2: Reaction scheme of salmeterol and impurities. EP is the European Pharmacopoeia. NaH is sodium hydride. TBAB is tetra-n-butylammonium bromide DMSO is dimethyl sulfoxide. NABH4 is sodium borohydride. Pd/C is palladium on carbon.
In the case of salmeterol, 4-phenyl butanol reacts with 1,6-dibromohexane to give Intermediate 1, which in turn reacts with benzylamine in the presence of dimethyl sulfoxide and triethylamine to yield N-[6-(4-phenyl butoxy)hexyl)] benzenemethanamine, a DMF starting material for salmeterol (see Figure 2). The compound 4-phenyl butanol is commercially available and prepared from benzene with succinic anhydride (7–11). If the benzene has a trace amount of toluene, the toluene is converted to 4-(4-methylphenyl)-1-butanol. The compound 4-(4-methylphenyl)-1-butanol is present in 4-phenyl butanol as a starting material impurity, which undergoes further reaction, similar to 4-phenyl butanol, to afford the methyl salmeterol impurity (see Figure 2). Similarly, the presence of 2-phenylethanol, 3-phenyl-1-hydroxypropane, and 4-phenyl-2-hydroxybutane in the 4-phenyl butanol will yield known salmeterol Impurities B, C, and E, respectively.
Similarly, 6-hydroxy and dichloro impurities, if present in the DMF starting material of ciprofloxacin, will be converted to European Pharmacopoeia impurity F and nonpharmacopeial impurity (chloro ciprofloxacin) at 2.1 RRT.
Intermediates
Organic compounds formed during the synthesis of APIs are termed as intermediates. The compound in the synthetic chain before the production of the final desired compound is called the penultimate intermediate.
Impurities due to rearrangement. Developing practical synthetic routes to render high-yield products in shorter stages or in a one- or two-pot reaction generally involves formation of rearranged intermediates that ultimately give the required final product.
As an example, the cyclization of bromonitrostyrene in the API ropinirole involves the rearrangement of the intermediate cyclic ion to give the indole ring with the formation of hydroxamic ester and chlorooxime acetate as impurities.
Impurities due to in situ reactions. Advances in synthetic chemistry have enabled a number of stages in a reaction to be carried out in just one or two pots without the need to isolate intermediates. The downside of such reactions is the unexpected and numerous impurities that form because intermediates and reagents are not isolated.
As an example, the alkylation of the key starting material (S)-2-amino butyramide for the API levetiracetam with chlorobutyrylchloride using potassium hydroxide in the presence of tetra-n-butylammonium bromide gives an intermediate that eventually cyclized into levetiracetam. This intermediate, however, is present in the final product as an USP impurity A.


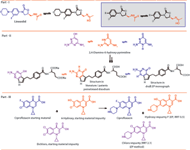
Figure 3: Linezolid (e.g., oxazolidinones class) and pemetrexed disodium tautomer impurity. EP is the European Pharmacopoeia. RRT is relative retention time.
Nonreactive intermediates. Nonreactive intermediates are impurities formed in some intermediate stage by the reaction of reagents used in the next stages due to carryover. Such impurities remain nonactive in the later stages.
For example, 4-phenyl butanol is a key raw material for the synthesis of salmeterol Intermediates 1 and 2 (see Figure 2). Intermediate 1 reacts with 4-phenyl butanol in the presence of sodium hydride and toluene to yield Compound 1, which is a nonreactive impurity in further stages. Intermediate 2 reacts with the trace amounts of Intermediate 1 and in the same conditions react to form Compound 2 (see Figure 2).
Reactive intermediates. Reactive intermediates, as the name implies, are byproducts or impurities resulting from the intermediate stages of the reaction that have the potential to react with the reagents or catalysts used in later stages. They are carried forwarded in every stage up to the final API as a reactive intermediate.
During the process development of salmeterol, an unknown impurity was detected at 2.08 RRT at a level of 0.11% and later identified after isolation to be Compound 3 (see Figure 2). The impurity formed in the final API due to presence of N-benzyl-6-(4-cyclohexylbutoxy)hexan-1-amine in Intermediate 2 leads to the salmeterol cyclohexyl impurity (12).
The reactive intermediate, N-benzyl-4-phenylbutan-1-amine is present in Intermediate 2 (see Figure 2). It is formed by the reaction of 4-phenyl butanol with benzyl amine and competes in all reaction stages with Intermediate 2 to form Compound 4 (see Figure 2).
A main challenges faced in developing the olefination route of the API aprepitant was a subsequent reaction of the vinyl ether intermediate with dimethyltitanocene to form an ethyl impurity (13).
Bis-compound impurities. The formation of new or unknown impurities can occur when scaling up a process, even with successful runs at a smaller scale. Examining the molecular weight of such impurities often reveals the compound is exactly double the weight of that being formed in that reaction step. Such dimeric derivatives are called bis-compound impurities. Two bis-compound impurities were formed in the intermediate and final stages in the synthesis of linezolid, to be discussed in Part III of this article.
Byproducts
In synthetic organic chemistry, getting a single end product, 100% pure, seldom occurs because of the change into byproducts, which can be formed through a variety of side reactions, such as incomplete reactions, overreactions, isomerization, or unwanted reactions between starting materials, intermediates, chemical reagents, or catalysts. For example, in the bulk production of paracetamol, diacetylated paracetamol may form as a byproduct (14).
In the Claisen rearrangement of the aryl propargyl ether in diethylaniline at elevated temperatures, formation of the desired chroman product is accompanied by the generation of a furan byproduct in success sively increasing amounts (15).
In the ropinirole synthesis, a somewhat similar case is observed in the final step. The reaction between the ropinirole precursor 4-(2-bromoethyl)-13-dihydro-2H-indol-2-one and di-n-propyl amine in water produces ropinirole in modest yield (57%), together with styrene as the major byproduct (38%) (16).


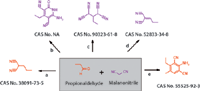
Figure 4. Propionaldehyde with malanonitrile reactions. CAS refers to Chemical Abstracts Service, No. is number, and NA is not available. Conditions: (a) with piperidine in pyridine, heating (Ref. 27); (b) with piperidine in pyridine, heating, cyclization (Ref. 28); (c) with piperidine, 1,4-dioxane (Ref. 29â30); (d) With [C4DABCO][BF4] in water, Time = 0.0166667 h, T = 20 °C, Knoevenagel condensation or with aluminum oxide in dichloromethane, T= 20 °C, Knoevenagel condensation aldol-condensation (Ref. 31â33).; and with morpholine in ethanol, T = 20 °C, Knoevenagel condensation (Ref. 34â37).
In another example, thiophenes are important heterocyclic compounds that are widely used as building blocks in many agrochemicals and pharmaceuticals (17). The synthesis of 2-amino-5-methylthiopene-3-carbonitrile is achieved by reacting a mixture of sulfur, propionaldehyde, malononitrile, and dimethylformamide using triethylamine (18–26).
The reaction of propionaldehyde with malononitrile and sulfur resulted in formation of two unknown impurities up to 7%, which were isolated and confirmed by 1 H NMR (nuclear magentic resonance spectroscopy), correlation spectroscopy, nuclear Overhauser effect spectroscopy, and single X-ray crystallography to be Impurity 1 (see Figures 4 and 5). These impurities are further found to react with 2-fluoro nitrobenzene to give next-stage impurities and which are controlled by purification in the respective stages.
Impurity 1 (see Figure 5) is a novel tricarbonitrile bicyclic compound, and as of the writing of this article, it is not known in the literature. Prediction of cLogP is 0.65, drug linkness is 4.04, and the drug score is 0.45 as determined by OSIRIS Property Explorer, software used to calculate various drug-relevant properties of chemical structures. Structure–activity relationship, quantitative structure–activity relationship, and drug design with other modified organic/inorganic hetrocyclic moieties could give some biological activity. The molecular designing of Impurity 1 for specific and unspecific purposes (e.g., DNA-binding, enzyme inhibition, anticancer efficacy) is based on the knowledge of molecular properties, such as the activity of functional groups, molecular geometry, and electronic structure, and on information cataloged on analogous molecules. The compound 2,6-diamino-7-ethyl-8-methylbicyclo[2.2.2]octa-2,5-diene-1,3,5-tricarbonitrile could be coupled with an active or nonactive peptide to check the biological activity as a prodrug or drug. The potential therapeutic and prophylactic activities of antimalarials, antimitotics, and antitumor agents could also be performed. This bicyclic compound may be used alone as a single agent or in combination with any organic or inorganic salts in chemotherapy or in combination with other chemotherapeutic agents after in vivo and in vitro testing.



Figure 5: Reaction scheme of olanzapine impurities. DMF is dimethylformamide. TEA is triethylamine. Addn is addition. RT is room temperature. CAS is Chemical Abstracts Service, No. is number, and NA is not available.
Transformation products
Transformation products deal with theorized and nontheorized products produced in a reaction. They can be synthetic derivatives of byproducts and are closely related to byproducts.
A reaction where transformation products occur is the formation of chloro acetyl derivative of salicylaldehyde during the acylation reaction of salicylaldehyde with bromo acetyl bromide using methylenedichloride (MDC) and aluminum chloride (AlCl3). Mechanistically, the formation of chloroacetyl derivative using bromoacetyl bromide could not be expected, but hypothetically, it could occur as a transformation reaction due to halogen exchange. During Friedel–Craft acylation with Lewis acid AlCl3 in methylene dichloride, the Lewis acid forms an ionized complex [Cl–AlCl2–Br]–, which eventually undergoes halogen exchange with the bromo acylium ion to yield the chloro acetyl derivative. Formation of this impurity in reaction is as high as 7–20%, which is an uncontrolled impurity in the manufacturing process. Nevertheless, this impurity would not affect the purity of the final drug substance because the reaction of the transformed impurity with 2 (see Figure 6, Part I) forms the desired product, salmeterol. The presence of the chloro impurity also has been confirmed by experiment (see Figure 6, Part II).



Figure 6: Chloro impurity-formation scheme of salmeterol. HPLC is high-performance liquid chromatography. MDC is methylenedichloride; AlCl3 is aluminum chloride.
Interaction products
The term interaction product deals with the interaction of two or more intermediates/compounds with various chemicals, intentionally or unintentionally. An interaction product is slightly more comprehensive than byproducts and transformation products. Two types of interaction products that are commonly encountered are drug substance–excipient interactions and drug substance–container/closure interactions.
Related products
The term related products means that the impurity has similar structure as that of the drug substance and may exhibit similar biological activity. This structural similarity by itself, however, does not provide any guarantee of similar activity. An example of a related product is 8-fluoro olanzapine.
Degradation products
Impurities formed by decomposition or degradation of the end product during manufacturing of the bulk drug are called degradation products. The term also includes degradation products resulting from storage, formulation, or aging. Parts II and III of this article will discuss the types and sources of the degradation products in further detail.
Tautomer impurities
Tautomers are readily interconvertible constitutional isomers that coexist in equilibrium. For APIs or drug molecules that exhibit tautomerism, there has been a confusion in identifying the two tautomeric forms. If one tautomer is thermodynamically stable and is the major form, the other tautomer should be considered as an impurity or simply termed as a tautomer of the API or drug molecule. To the best of the authors' knowledge, there has been no literature relating to the isolation, synthesis, or characterization of a tautomeric impurity(-ies) from the final API.
Linezolid is an treatment for nosocomial infections involving gram-positive bacteria. Oxazolidinones possess a unique mechanism of bacterial protein synthesis inhibition (38–39). Linezolid has an N-acetyl group (–NH–CO–CH3) due to that lactam–lactim tautomerism, which may occur during the synthesis but also may be stable. An effective analytical method needs to be developed to identify both tautomers.
A key starting raw material of pemetrexed disodium 2,4-diamino-6-hydroxy-pyrimidine shows the keto-enol form occurring in different ratios and which will be converted to the final drug using a known synthesis (see Figure 3).
Tautomers vary in their kinetic and thermodynamic stability, thereby making it difficult to determine whether they could be separated, isolated, or analyzed. Keeping this in mind, the use of the term impurity for tautomers in a final API/drug moiety presumably will be an important discussion in near future.
Conclusion
Part I of article highlights the origination and classification of impurities and provides a perspective on impurities in drug substances and drug products. The impurity profile of a drug substance is on increasing importance for ensuring the quality of drug products. Whatever the class of impurity, its identification and adequate control is a tremendous challenge for process-development chemists. Because no two drugs are alike, neither are two development pathways. Each drug candidate poses a different challenge in terms of impurities, and establishing efficient ways for the isolation and control of impurities is a key task in process development.
Part II of this article, to be published in the March 2012 issue of Pharmaceutical Technology, will discuss chiral and polymorphic impurities. Part III, to be published in the April 2012 issue of Pharmaceutical Technology, will discuss genotoxic and stability impurities.
Kashyap R. Wadekar, PhD,* is a research scientist (II), Mitali Bhalme, PhD, is an associate research scientist, S. Srinivasa Rao is a research associate, K. Vigneshwar Reddy is a research associate, L. Sampath Kumar is a research chemist, and E. Balasubrahmanyam is a research chemist, all with Neuland Laboratories, 204 Meridian Plaza, 6-3-854/1, Ameerpet, Hyderabad, India, tel. 91 40 30211600, kashyapwadekar@neulandlabs.com.
*To whom all correspondence should be addressed.
Submitted: Sept. 19, 2011; Accepted Nov. 28, 2011.
References
1. FDA, Guideline for Submitting Supportive Documentation in Drug Applications for the Manufacture of Drug Substances (Rockville, MD, Feb. 1987).
2. ICH, Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients, Step 5 (Nov. 2000).
3. FDA, Draft Guidance for Industry: Drug Substance: Chemistry Manufacturing and Controls Information (Rockville, MD, Jan. 2004).
4. T. Cupps et al., Pharm. Technol. 27 (2), 34–52 (2003).
5. M. Johnson, Med. Res. Rev. 15 (3), 225–257 (1995).
6. A.T. Nials et al., Am. Rev. Resp. Dis. 149, A481 (1995).
7. Y Kawakami et al., Eur. J. Med. Chem. 31 ( 9), 683–692 (1996).
8. N.O. Mahmoodi and M. Jazayri, Syn. Comm.31 (10), 1467–1476 (2001).
9. M. Islam et al., Acta Poloniae Pharm. Drug Res., 65 (4) 441–447 (2008).
10. K.T. Chapman et al., Bioorg. Med. Chem. Lett. 6 (7), 803–806 (1996).
11. A.A. Siddiqui et al., Bioorg. Med. Chem. Lett. 21 (3), 1023–1026 (2011).
12. B. Venkatasubbaiah et al., Scientia Pharm. 77, 579–587 (2009) .
13. J.J. Hale et al., J. Med. Chem. 41 (1), 4607–4614 (1998).
14. K.M. Alsante, Amer. Pharm. Rev. 4 (1) 70–78 (2001).
15. J. Zsindely et al., Helv. Chim. Acta 51, 1510 (1968).
16. J.D. Hayler et al., Org. Process Res. Dev. 2 (1), 3–9 (1998).
17. J. Swanston, "Thiophene" in Ullmann's Encyclopedia of Industrial Chemistry, (Wiley-VCH, Weinheim, Germany, 2006).
18. Tel-Aviv University, "Novel Psychotropic Agents Having Glutamate NMDA Activity," WIPO Patent WO2008/50341, May 2008.
19. Watson Pharmaceuticals, "2-Methyl-thieno-benzodiazepine Process," WIPO Patent WO2004/94390, Nov. 2004.
20. Shastri et al., "Process for Producing Pure Form of 2-Methyl-4-(4-Methyl-1-Piperazinyl)-10H-Thieno[2,3-b] [1,5]Benzodiazepine," US Patent 2009/5556, Jan. 2009.
21. Eli Lilly, "Process for Preparing 2-Methyl-thieno-benzodiazepine" US Patent 6008216, Dec. 1999.
22. Lilly Industries, "2-Methyl-thieno-benzodiazepine," US Patent 5229382, July 1993.
23. Eli Lilly, "2-Methyl-thieno-benzodiazepine," US Patent 5605897, Feb. 1997.
24. X He et al., J. Pharm. Sci. 90 (3) 371–388 (2001).
25. V.P. Shevchenko, Russian J. Bioorg. Chem. 31 (4), 378–382 (2005).
26. V.P. Shevchenko, Bioorganicheskaya Khimiya 31 (4) 420–424 (2005).
27. J.C. Dunham et al., Synthesis, 4, 680–686 (2006).
28. A.H. Elgandour et al., Indian J. Chem. Sec. B: 36 (1) 79–82 (1997).
29. R. Mariella and A. Roth, J. Org. Chem. 22 (9), 1130 (1957).
30. Hart and Freeman, Chemistry and Industry, p. 332 (1963).
31. Da-Zhen Xu et al., Green Chem. 12 (3) 514–517 (2010).
32. H.C. Brown and M.V. Rangaishenvi, J. Heterocycl. Chem. 27 (1), 1–12 (1990).
33. S. Fioravanati, Synlett. (6), 1083–1085 (2004).
34. V.D. Dayachenko, J. Gen. Chem. 74 (7), 1135–1136 (2004).
35. Zhurnal Obshchei Khimii 74 (7), 1227–1228 (2004).
36. V.D. Dayachenko and A.N. Chernega, Russian J. Org. Chem. 42 (4), 567–576 (2006).
37. Zhurnal Organicheskoi Khimii 42 (4), 585–593 (2006).
38. D.L.K. Marotti et al., AntiMicrob. Agents Chemother. 41 (10), 2132–2136 (1997).
39. E.Z. Gray et al., Expert Opin. Investig. Drugs 6 (2), 151–158 (1997).





")






Newsletter
Get the essential updates shaping the future of pharma manufacturing and compliance—subscribe today to Pharmaceutical Technology and never miss a breakthrough.














Drug Solutions Podcast: A Closer Look at mRNA in Oncology and Vaccines
April 30th 2024In this episode fo the Drug Solutions Podcast, etherna’s vice-president of Technology and Innovation, Stefaan De Koker, discusses the merits and challenges of using mRNA as the foundation for therapeutics in oncology as well as for vaccines.



