Defects in Herceptin vials stem from a fault in the packaging process.
Quality Systems
Latest News
Advertisement
Advertisement

The production staff was sure the lab couldn't test their way out of a paper bag.
Pharmaceuticals current good manufacturing practice (CGMP) violations accounted for just 36 of the 441 Warning Letters issued by the US Food and Drug Administration in 2006.

Symposium held at 2006 AAPS Annual Meeting in San Antonio
US District Court for the Eastern District of New York Magistrate Judge A. Kathleen Tomlinson's Nov. 30 2006 "Report and Recommendation" supporting a preliminary injuction requested by the plaintiffs in RxUSA Wholesale, et al v. FDA.
Washington, DC (Dec. 13)-After considerable debate and negotiation, Congress this week passed four bills poised to affect pharmaceutical and biotechnology research, development, and manufacture. All are currently awaiting signature by the President.
Rockville, MD (Dec. 12)-The US Food and Drug Administration is proposing amendments to its final rule regarding labeling requirements for convenience-size over-the-counter human drugs.
Dec. 11, 2006 (Rockville, MD)-Effective Jan. 16, 2007, Scott Gottlieb, MD, deputy commissioner for medical and scientific affairs at the US Food and Drug Administration, will leave the agency and will return to the American Enterprise Institute.
Rockville, MD ( Dec. 4)-The U.S. Pharmacopeia plans to open a 10,500-square-foot site in Shanghai in February 2007. The facility, in Shanghai's Zhangjiang Hi-Tech Park, will support collaborative testing, technical assistance, customer service, and training.
Washington, DC (Dec. 7)-One of the last acts of outgoing Senate majority leader Bill Frist (R-Tenn) was to push through confirmation of Andrew von Eschenbach as the official head of the Food and Drug Administration.

Brand-name manufacturers seek to protect their products.

Poor support of drug- and process-development training programs will affect industry's future growth.
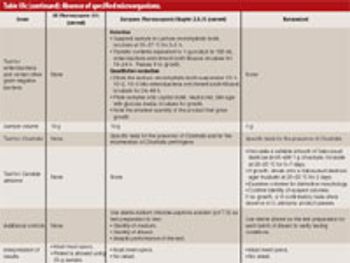
The US Pharmacopeia (USP), the Japanese Pharmacopoeia, and the European Pharmacopoeia "Microbial Limits Tests" are in the final stages of harmonization. The harmonized USP chapters are slated for implementation in 2009. This article describes the harmonized USP Chapters <61> "Microbial Enumeration," <62> "Absence of Specified Microorganisms," and <1111> "Microbiological Attributes of Nonsterile Pharmaceutical Products," and suggests they will likely require some revalidation of existing methodologies. Companies should put plans in place immediately for this work and show consistent progress towards this goal.

We really knew what we were doing-until we opened the column for a routine repacking.
Proof of concept is a new development paradigm.

EFPIA's 'Mock P.2' document aims to show how the role of 'quality risk management' and process analytical technology as an enabler for quality by design can be presented in a common technical document format. This article summarizes the main features of this document, and explains the key concepts and principles used.
London (Nov. 22)-The European Medicines Agency reports a defect in some vials of Herceptin (trastuzumab), the anticancer treatment by Roche, which have been distributed in Europe. As a result, The EMEA's Committee for Medicinal Products for Human Use outlined a plan, formulated in conjunction with Roche, for the visually reinspecting and replacing defective vials.
Basel, Switzerland (Nov. 17)-Novartis plans to submit additional information to the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency on manufacturing issues to support the approval of its antifungal drug "Mycograb." The CHMP issued a negative recommendation for the drug.
Washington, DC (Nov. 13)-The Biotechnology Industry Organization is urging the World Health Organization to assign a distinct international nonproprietary name (INN) to each unique biological product, including follow-on biologics or biosimilars.
Over-the-counter drug maker Perrigo Company issued a voluntary recall of certain lots of store-brand acetaminophen 500-mg caplets that could contain trace amounts of metal particulate. Approximately 11 million bottles of acetaminophen are affected by the recall.
Mystic, CT (Oct. 26)-Kenneth G. Chapman died at his home here on Oct. 26. He was 79. The cause was pancreatic cancer
Dallas, TX (Nov. 7)-The US Food and Drug Administration has posted a Warning Letter from its Dallas District office to Matheson Tri-Gas, Inc.
AAPS, San Antonio (Oct. 31)-Industry and regulatory agency concerns over process variability have prompted both groups to take a closer look at analytical method transfer processes.
AAPS, San Antonio (Oct. 31)-Excipient manufacturers are raising concerns over recently adopted European guidelines, set to become effective January 1, 2007, which provide a framework and approach for dealing with genotoxic impurities in new active substances.
Liverpool, UK (Oct. 20)-Novartis Vaccines and Diagnostics Limited is recovering two lots totaling 500,000 doses of its ?Fluvirin? influenza virus vaccine after reports that the product had been received from distributor Cardinal Health Care in either a frozen state or below the required storage temperature range of 35?46 degrees F.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
FDA Grants Priority Review For Pfizer’s Marstacimab for Hemophilia A or B
2
PharmTech Weekly Roundup - February 6, 2026
3
Rare Disease Treatments: Navigating the Economics of Global Innovation
4
Hovione’s Strategy for Complexity, Speed, and Regional Supply Chains
5