
Pharmaceutical Science & Technology News
Drug Development
Latest News
Advertisement
Research into Cold Cancers Heating Up
Accurately targeted immunotherapies through reliable neoantigen recognition enable personalized medicine development.
Development of Gamma-Delta T-Cell Therapies
Activation and expansion are essential for success in both autologous and allogeneic therapies.
QbD for Small-Molecule Continuous Process Development
Continuous manufacturing and a quality-by-design development approach are a natural fit.

Advertisement
In part two of a series of three articles, mixing rates and mechanisms are examined using rectangular bin blenders and two free-flowing mixtures.
The limitation-of-risks (LR) method can be used as an engineering tool in risk assessment work for the identification, minimization, and evaluation of potential airborne risks, and for the identification of adequate monitoring points.
The authors outline stability studies designed to evaluate the effect of temperature excursions on product quality that may occur during distribution.
The role of microbial testing to ensure the sterility of aseptically filled sterile products is explained, from the product development phase to in-process monitoring to finished product testing.
Pharmaceutical Science & Technology News
A recent USP gathering generated creative suggestions for the March 2005 USP convocation.
pharmaceutical science and technology news
Added functionality excipients facilitate the development of novel drug delivery methods and improve processing techniques.
Currently, high-production rates and continuous production processes favor existing tableting technologies. However, if tablet development becomes rate-limiting in the future, alternative technologies may prove attractive.
Some of the common problems faced by formulators and how using ion exchange resins may be able to solve them are discussed.
Recent advances in spray-drying technology have led to the production of new directly compressible lactose grades with distinct advantages.
Recent technology improvements have made acrylics the preferred system for the aqueous enteric coating of tablets.
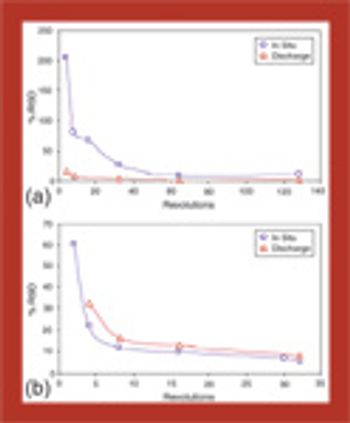
In this series of articles, bin blender performance is comprehensively reviewed using both free-flowing and cohesive mixtures. In part 1, an introduction to tools and techniques is presented, followed by an examination of parameter effects, mixing mechanisms, and the effects of cohesion on mixing.
The author describes a separation method for two active ingredients in the contraceptive pill with liquid chromatography UV detection.
The author suggests that an excipient's functionality can only be determined in the context of a specific formulation and manufacturing process.
Bin blender performance is comprehensively reviewed using both free-flowing and cohesive mixtures.
Pharmaceutical Science & Technology News

Pharmaceutical Technology Europe
FDA expects a firm that is subject to GxP to develop a risk evaluation of its product and to then mitigate the identified risks. Identified risks may be addressed by technical fixes that effectively eliminate the risks or reduce the likelihood of occurrence and/or severity of consequences to acceptable levels.
Current microbiological methods cannot measure microbial contamination at the levels that engineers and regulators seek to establish for aseptic processing cleanrooms. New approaches for assessing data and establishing alert and action levels are advocated, and an example of one analytical tool is considered.
Synthetic excipients frequently offer advantages over all-natural compounds.
The authors investigate whether an extrusion-spheronization proces can be used to develop a matrix-based, controlled-release formulation for a highly water-soluble drug.

Pharmaceutical Technology Europe
Dry powder inhalers are a well-accepted dosage form for pulmonary drug delivery and a wide variety are either currently available or in development. This article examines a premetered, capsule-based multidose inhaler for which different qualities of a-lactose monohydrate were screened.

Pharmaceutical Technology Europe
The bioavailability of some insoluble drugs is enhanced when they are dissolved in the solubilizing agent macrogol 400, although conventional hard capsules cannot tolerate the agent. This article investigates a PVA copolymer, which has been developed by the authors, examining its properties and its suitability as a material in capsule formulations.

Pharmaceutical Technology Europe
Following the launch of its initiative, "Pharmaceutical cGMPs for the 21st Century: A Risk-Based Approach," FDA has been looking to process analytical technology (PAT) for improvements in process efficiency and quality. This article discusses the implementation of PAT systems into production environments, its impact on quality assurance and the necessity of an integrated approach. Options for implementing PAT are also presented.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Seventeen Peptide Guidance Revised: What Developers Need to Know
2
GSK Cambridge Move Signals Shift in Drug Scale-Up Strategy
3
Midyear 2026 Industry Report: Taking Stock of Pharma Manufacturing, AI, and Regulatory Trends
4
Pharma Fundamentals: An Introduction to Hold Times in Biopharmaceuticals
5