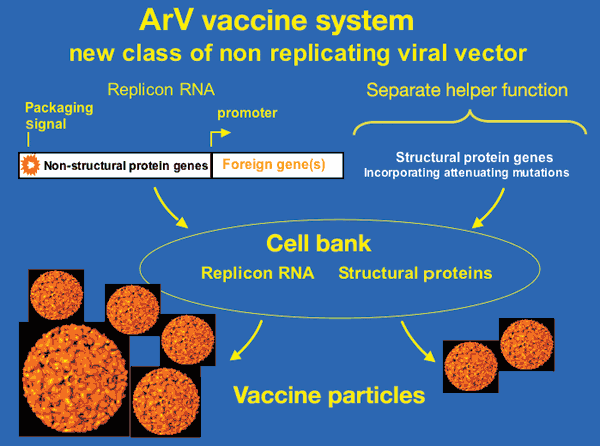
Vaccine developers are wrking on new drug delivery systems that offer improved immune responses, better stability, and a wider pool of targeted diseases.
Drug Development
Latest News
Advertisement
Research into Cold Cancers Heating Up
Accurately targeted immunotherapies through reliable neoantigen recognition enable personalized medicine development.
Development of Gamma-Delta T-Cell Therapies
Activation and expansion are essential for success in both autologous and allogeneic therapies.
QbD for Small-Molecule Continuous Process Development
Continuous manufacturing and a quality-by-design development approach are a natural fit.

Advertisement
The authors review the solution and powder properties of polyethylene oxide and its various applications in hydrogels and hydrophilic matrix systems.
The unit-dose bar coding rule requires integrating many aspects of packaging design and control.
FDA is finalizing guidances and enforcing compliance with manufacturing standards as part of its efforts to ensure drug safety.
A newly developed software program transforms written SOPs for all required analytical method validation experiments into transferable automated templates, integrating individual activities and technologies under one platform.
Major pharmaceutical companies are focusing more than ever on the prices CROs are charging for their services.
New technologies and improvements to existing ones can reduce contamination risk in aseptic processing.
A ready-to-fill closed vial can improve aseptic filling quality and reduce process complexity.
The complete elimination of human-derived contamination is possible only with the elimination of human intervention.
Pfizer Recalls Lot of Neurontin
Crucell and NIH Sign Ebola Vaccine Manufacturing Contract
The Empire Strikes Back: Innovators Releasing Generic Drugs
Warning Letter: Compounding at Palace Pharmacy
The authors propose a strategy for classifying and validating inprocess testing methods.
The author proposes a new analytical graphic, the sector chart, which presents data that cannot be adequately presented with current graphs.This chart combines features of zone charts with the basic principle of precontrol charts. It addresses engineering control and is superior for representing data such as beneficial and adverse trends.
The validation of alternative microbiological testing is an opportunity for a manufacturer to decrease the amount of time required for laboratory results.To properly validate these alternatives, a practitioner must first identify what is being studied. The regulatory effect on established product and process specifications and levels must be completely evaluated, as changing the method of analysis may well change the pparent number in the sample.
RFID and battery-powered containers are among the developments receiving attention in the pharmaceutical cold chain.
X-ray microtomography has great potential for improving the understanding of the structural features of solid dosage forms and the changes in those features during manufacturing, handling, and storage.This article describes the basic principles of the technique and provides examples of its potential applications.
The sterility testing of samples from an aseptic process may be considered an entirely separate aseptic process that is subject to the same types of adventitious contamination as the aseptic process itself.
A comparative study of three air samplers used for bioaerosol collection was performed to evaluate the average recovery of colony-forming units and assess the precision of each device.
What the industry needs most is not reduced oversight. What we need is, competent, constructive, informed, consistent, fair, and predictable inspection.
To meet the requirements of the USP ^755& Minimum Fill and ^698& Deliverable Volume tests, target fill levels greater than 100% must be established.This article proposes a criterion for establishing an appropriate target fill level such that a sample will have a 95% probability of passing these USP tests at 95% confidence.
The authors argue that chlorine dioxide (CD) is a safe and effective decontaminating agent that can be used for challenging applications.The effectiveness of CD gas for sterilizing complex isolator systems is studied.
Pharmaceutical Technology Europe
Nanotechnology is believed to hold enormous promise for the future of medicine and healthcare...
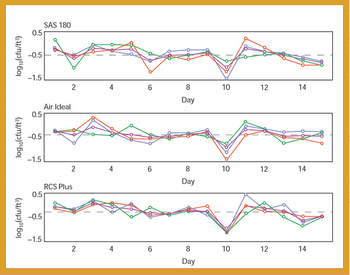
A comparative study of three air samplers used for bioaerosol collection was performed to evaluate the average recovery of colony-forming units and to assess the precision of each device.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Seventeen Peptide Guidance Revised: What Developers Need to Know
2
GSK Cambridge Move Signals Shift in Drug Scale-Up Strategy
3
Midyear 2026 Industry Report: Taking Stock of Pharma Manufacturing, AI, and Regulatory Trends
4
Pharma Fundamentals: An Introduction to Hold Times in Biopharmaceuticals
5