FDA is finalizing guidances and enforcing compliance with manufacturing standards as part of its efforts to ensure drug safety.
Quality Systems
Latest News
Advertisement
Advertisement

Pharmaceutical Technology Europe
The FDA initiative —Process Analytical Technologies (PAT) — is slowly gaining momentum, creating a revolution in manufacturing and testing processes that aims to ensure product quality. Its growth will encourage faster testing techniques to bring analytical testing closer to on- and at-line testing during the product manufacturing process.

Pharmaceutical Technology Europe
While Europe's political leaders were paying lip-service to creating a more competitive Europe at their spring summit in Brussels, European pharmaceutical technologists were tackling the serious business of turning political rhetoric into practical reality.
Study results show that the state of a cleanroom clothing system–new or much used–influences the protection efficacy of the system.
Pfizer Recalls Lot of Neurontin
Drug Recall
HHS Awards $97-Million Vaccine Development Contract
Warning Letter: Compounding at Palace Pharmacy
Pharmaceutical Science and Technology News
Managers of FDA-regulated firms must be proactive in how they manage their company?s compliance with good manufacturing practices regulations.
The C-SOC team must develop a network of committed industrial partners who will participate in the direction, execution, and evaluation of research and educational activities.
An increasing number of new compounds are being introduced into pharmaceutical pilot plants.The knowledge base for these compounds regarding their toxicities,physical handling, and cleaning is limited.The authors examine various approaches for addressing the cleaning validation of new compounds and discuss the role of determining appropriate visible residue limits.
At its 2005 Convention, USP passed 13 resolutions that will help define the organization’s path for the next five years and represent the interests of its diverse constituency.
Innovators and generics makers are staking out positions on follow-on proteins and other manufacturing issues.

Managers of FDA-regulated firms must be proactive in how they manage their company's compliance with good manufacturing practices regulations.
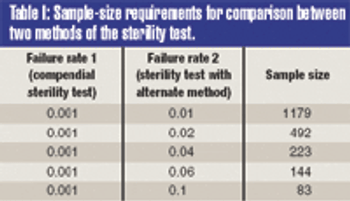
The validation of alternative microbiological testing is an opportunity for a manufacturer to decrease the amount of time required for laboratory results. To properly validate these alternatives, a practitioner must first identify what is being studied. The regulatory effect on established product and process specifications and levels must be completely evaluated, as changing the method of analysis may well change the apparent number in the sample.
FDA Finalizes Guidances on Minimizing Drug Risk and Submitting Pharmacogenomic Data
FDA New Drug Review Times Continue to Drop in 2004
USP to Publish Spanish Translation of USP-NF; Council of Experts Named
Warning Letters: DanChem Technologies & Medsep
US Marshals Seize Supplies of GSK Paxil CR, Avandamet
UK MHRA Okays Chiron Flu Vaccine Plant; FDA Approval Still to Come
FDA Approves Treatment for Smallpox Vaccination Side Effects
pharmaceutical science and technology news
IPEC-Americas has just completed a major update to its significant change guideline to address current issues in the manufacture of excipient ingredients and to assist manufacturers in developing an impurity profile.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
FDA Activity Round Up – July 2026
2
Midyear 2026 Check-In: AI, Supply Chains, and the NDC-12 Story Nobody's Watching
3
Can Governance-by-Design Make AI Compliant in Pharma? (Part II)
4
From Predictions to Reality: Mike Stenberg Assesses Pharma Manufacturing's First Half of 2026
5