On the other hand, if we don't keep up the pressure on counterfeiting in the US, we may share the plight of the developing world, where nearly 25% of all drugs sold are fakes.
Quality Systems
Latest News
Advertisement
Advertisement

Pharmaceutical Technology Europe
The collection, investigation and monitoring of suspected adverse drug reactions (SADRs), and associated product use and complaint information, is a regulatory requirement for all manufacturers of pharmaceuticals for human use.1 This process, called pharmacovigilance or drug safety, appears to be fairly standardized between different pharmaceutical companies and usually contains the elements outlined in Figure 1.

Pharmaceutical Technology Europe
Defenders of the research-based industry are hoping for an early Christmas present from the European Court of Justice (ECJ). All the signs are that a small but significant victory is on the way against parallel importing. Right at the end of October, a senior judge responsible for a leading case at the court made clear his view that international drug firms do not necessarily have to make life easy for parallel importers. The formal ruling on the case at issue is expected within weeks.
HHS
This article provides guidance for minimally acceptable method validation practices and a foundation for assessing the risks and benefits associated with method validation programs.
Pharmaceutical Science & Technology News
Phase II of FDA's GMP modernization plan may involve revising regulations to fit new policies and inspection and review programs.
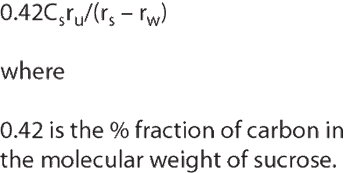
Pharmaceutical Technology Europe
The implementation of a total organic carbon (TOC) method into the United States Pharmacopeia (USP) has its origins in the early 1990s, when the Water Quality Committee (WQC) of the Pharmaceutical Manufacturers Association (PMA, later renamed PhRMA) debated improvements in the testing of purified water (PW) and water-for-injection (WFI). The resulting inclusion of modern analytical techniques replaced much older methods - some of which had been listed in the USP for more than 150 years. Finally, two new regulations were put in place: Chapter for conductivity, which replaced a series of individual ion tests; and Chapter which replaced the oxidizable substances test with a TOC method.
Stakeholders consider ways to improve printed patient inserts.
FDA and industry discuss proposals for regulating combination diagnostics, medical devices, and pharmacogenomic-based drug products.
Pharmaceutical Science & Technology News
Evaluations have shown, in most cases, visual observations are sensitive enough to verify equipment cleanliness. An experiment was conducted to explore the possibility of using a visible-residue limit as an acceptable cleaning limit in a pharmaceutical research facility, including an evaluation of the limits and subjectivity of ?visually clean? equipment.
The authors dispel common misinterpretations of the United States Pharmacopeia.
This article discusses the basics of sterile filter qualification and validation, with emphasis on bacterial challenge protocol development and testing. Reference is made to Technical Report no. 26 of the Parenteral Drug Association.
Pharmaceutical Technology Europe
FDA's recently released initiative has made process analytical technology (PAT) a hot topic in the life science industry. PAT describes the application of process analytical chemistry tools, feedback process control, information management tools, and product and process optimization strategies for the development and manufacture of pharmaceuticals. In this article, the author explores the impact PAT will have on the pharmaceutical industry.
FDA is re-engineering the CMC review process for innovator and generic drugs and backing risk-based ICH quality standards.
Pharmaceutical Science & Technology News
A straightforward approach reveals how the probability of passing the USP content uniformity test can be calculated for tablets and capsules.
Particulate generation and durability concerns are encouraging pharmaceutical manufacturers to seek alternatives to wood pallets.
Pharmaceutical Technology Europe
Customers are placing high demands on pharmaceutical manufacturers to produce sensitive, cost-effective and regulatory compliant inspection technology. This article examines how metal detection equipment suppliers can combine forces with manufacturers to provide state-of-the-art inspection systems, giving particular consideration to cleaning processes and regulatory issues.

Pharmaceutical Technology Europe
This article investigates what defines a best-in-class QC lab based on experience of implementing operational improvement projects in world-class labs. It includes an assessment of a benchmarking process, a case study of improvements made as a result at one company and findings on what constitutes best-in-class for QC labs.
Pharmaceutical Technology Europe
In Europe ... the onward rush of time has imposed no change at all during the last 25 years. Anyone could step back in at any point, at any moment, and find the same depressingly familiar debates, interminably failing to reach any real conclusions.
The author describes the efforts of the standards committees to establish guidelines for PAT implementation and the impact of the guidelines on the industry.
Pharmaceutical Science & Technology News
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Can Governance-by-Design Make AI Compliant in Pharma? (Part II)
2
Brent Wilhelm on Why Drug Shortages Persist Despite Available Inventory
3
Can AI Fix Pharma's Last-Mile Delivery Blind Spot?
4
The Implications of the FDA’s New Finalized Guidance Documents
5