
Misalignment around liquid filtration requirements and contamination control assurance still persist despite the revisions to Annex 1.
Misalignment around liquid filtration requirements and contamination control assurance still persist despite the revisions to Annex 1.

Contextualizing what type of flexibility is needed is paramount when considering cleanroom design.

A task force established by PDA and BioPhorum is establishing scientific data for integrity test activities.

Consider a matrix of factors when choosing what type of cleanroom facility to construct.

Why shouldn’t biopharmaceutical manufacturers be able to leverage standardized construction practices to improve time to market?

This article examines the changes now taking place, and what the future might bring.

Total cost of ownership, including operating costs, must be considered when deciding between flexible and traditional cleanroom systems.

Pharmaceutical Technology Europe
Biopharmaceuticals commonly cannot be terminally sterilised, as such aseptic processing using sterilising grade filtration is essential.

The authors describe the origins of single-use components and explain their application to aseptic processes. They also show how disposable devices have changed over time and offer a glimpse of the future.

The authors propose increased use of single-use technologies in biopharmaceutical manufacturing to achieve operational excellence without compromising product quality.
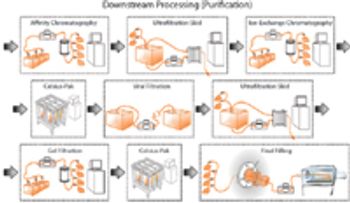
The authors discuss current and future disposable technologies and outline the validation and qualification steps that would be required for a possible disposable process stream.
Pharmaceutical Technology Europe
Although realized in 1983, ASTM's standrad test method for determining bacterial retention of membrane filters used for liquid filtration is still hugely beneficial to today's pharmaceutical industry.
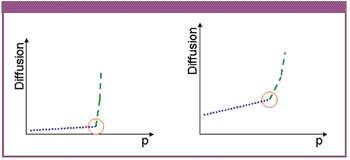
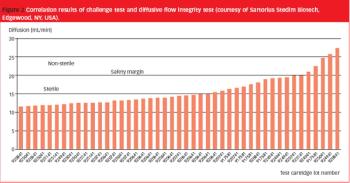
The authors describe a novel approach for the integrity testing of large sterile filter systems such as multiround housings and describe a multipoint diffusion test capable of detecting minor failures.
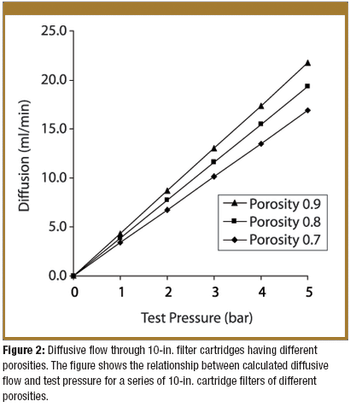
Pre-use integrity testing of sterilizing-grade filters eliminates the potential adverse effects of filter loading on the integrity-test results, allowing unambiguous correlation with the integrity-test specification established during filter-validation studies.
Pore-size ratings are so unrelated to actual dimensions and so subject to anomalous interpretations as to make substantial dependency upon their values an unwise choice. Moreover, the means of measuring them are questionable. The pore-size rating system at best provides a qualitative differentiation.

Model organisms are useful when validating sterile filtration, but successful retention of the model organism does not always guarantee that effluent is sterile. The authors explore the various factors that influence sterile filtration.

The authors encourage the investigation into whether the occurrence of grow-through and the diminution in the size of certain organisms when in contact with given liquids are the same phenomenon manifested under different circumstances.

Pharmaceutical Technology Europe
The regulating authorities have, it seems, a preference for the application of multimembrane combinations to maximize organism retentions.
This article critiques the diffusive-airflow and bubble-point tests for their comparative suitability for integrity testing in pharmaceutical processes.

Pharmaceutical Technology Europe
Sterilizing grade filters are widely used in the biopharmaceutical industry and were once thought of as being perfect. However, these filters have experienced rapid developments and improvements during the last decade, which have resulted in enhanced thermal and mechanical resistance. Moreover, their performance levels have been raised, which has led to significant cost savings within production processes.

January 2nd 2021

May 15th 2023

May 15th 2021

March 16th 2024

March 1st 2010

January 7th 2011