ePT--the Electronic Newsletter of Pharmaceutical Technology
Ranbaxy received full market approval from the US Food and Drug Admnistration for its anti-infective agent ?Clarithromycin? oral suspension.
ePT--the Electronic Newsletter of Pharmaceutical Technology
Ranbaxy received full market approval from the US Food and Drug Admnistration for its anti-infective agent ?Clarithromycin? oral suspension.
ePT--the Electronic Newsletter of Pharmaceutical Technology
President Bush signed the FDA Amendment Act into law, thereby reauthorizing the Prescription Drug User Fee Act (PDUFA).

As China emerges as a significant supplier of pharmaceutical ingredients, it must assure other countries of the safety of its excipients.

Amidst debate, the European Pharmacopoeia Commission is working to include physical or "functionality-related characteristics" of exipient materials in its monographs.

The United States House of Representatives passed its version of the Patent Reform Act of 2007 (H.R. 1908) in September in a 220-175 vote. Although the bill aims to fix current flaws in the US patent system and to bring it in line with those of other countries' systems, the biopharmaceutical industry is largely unhappy with the news, arguing that it will reduce patent protection.

To monitor and control processes or products, analytical methodology must be fit for purpose. An approach to apply quality by design principles to the design and evaluation of analytical methods has therefore been developed to meet these needs. This article features a downloadable template on which to conduct a failure mode effect analysis (FMEA).

Our files pull up lost contracts, poor competitive tactics, and an over-ambitious employee.

Regulators and industry professionals provide a guide for identifying and measuring impurities.

Courts are denying the terminally ill access to experimental drugs, leaving many with no options.

Offshore contracting is nothing new for the pharmaceutical industry. Neither is importing active ingredients, or prescription drugs, for that matter. But the authorities are having trouble keeping up with these growing international trends.

Orally disintegrating tablets (ODTs) continue to attract attention as an alternative to conventional oral dosage forms.

Expanded access to drugs for seniors has increased demand and focused attention on costs.
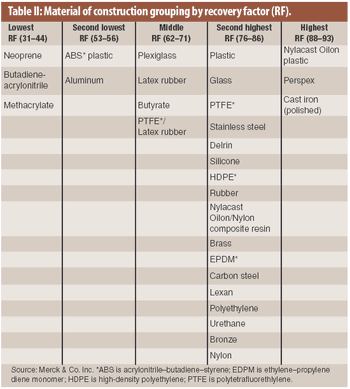
The material of construction is a factor in the recovery of residue in cleaning validation. An analysis of existing recovery data showed that recovery factors for drug products on various materials of construction may be categorized into several groupings.
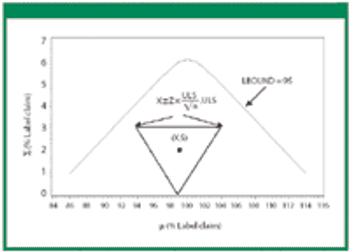
Revisions to the United States Pharmacopeia's (USP) uniformity test require manufacturers to establish new acceptance limits. The authors present their method for calculating acceptance limits consistent with USP's revised content-uniformity test requirements.

Pharmaceutical Technology Europe
The completion of the Human Genome Project in 2003 led to a flurry of predictions regarding the application of pharmacogenomics to drug development. With US and European regulatory authorities finally on the verge of issuing guidance on the use of pharmacogenomics, drug development is all set to change.
Pharmaceutical Technology Europe
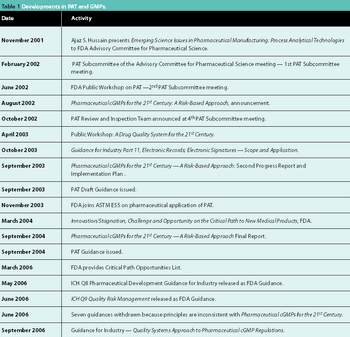
Recent regulatory initiatives have emphasized the need to improve pharmaceutical manufacturing. PAT marked the beginning of a number of regulatory efforts to encourage innovation and a transition towards science-based manufacturing. This article reviews the progress of the regulatory initiatives and describes two significant research initiatives to develop a future pharmaceutical manufacturing environment based on scientific understanding of pharmaceutical materials and processes.
ePT--the Electronic Newsletter of Pharmaceutical Technology
The European Medicines Agency?s Committee for Medicinal Products for Human Use recommended the lifting of the suspension of the marketing authorization for ?Viracept? (nelfinavir mesylate) from Roche and the re-introduction of the drug in the European Union.
ePT--the Electronic Newsletter of Pharmaceutical Technology
Rep. John D. Dingell (D-MI), chairman of the Committee on Energy and Commerce, along with Reps. Frank Pallone (D-MI), chairman of the Subcommittee on Health, and Bart Stupak (D-MI), chairman of the Subcommittee on Oversight and Investigations, introduced legislation that would create a user fee on imported drug and food shipments.
House and Senate leaders finally agreed on compromise legislation to renew prescription user fees, just a few days before the funding program was set to expire.
ePT--the Electronic Newsletter of Pharmaceutical Technology
US Food and Drug Administration Commissioner Andrew von Eschenbach warned agency employees last Friday that 2,000 layoff notices could be coming as early as Sept. 21.
Merck & Co.?s Isentress, the company?s new first-in-class AIDS drug, has received a recommendation for approval from FDA.
ePT--the Electronic Newsletter of Pharmaceutical Technology
Company and People Notes: Baxter and Halozyme Expand Relationship, Crucell Names COO, More.
ePT--the Electronic Newsletter of Pharmaceutical Technology
More than 100 people attended this week’s Regulatory Affairs Conference by the International Pharmaceutical Excipients Council of the Americas to discuss the latest trends and challenges in the excipient supply chain.
ePT--the Electronic Newsletter of Pharmaceutical Technology
The US Food and Drug Administration’s Interagency Working Group on Import Safety released a Strategic Framework based on a cost-effective, risk-based approach for ensuring the safety of products exported to the United States.
ePT--the Electronic Newsletter of Pharmaceutical Technology
The United States House of Representatives passed its version of the Patent Reform Act of 2007 (H.R. 1908) in a 220–175 vote.