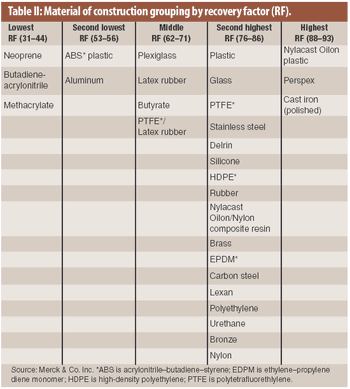
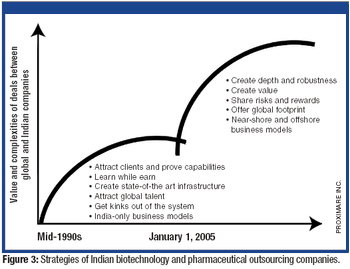
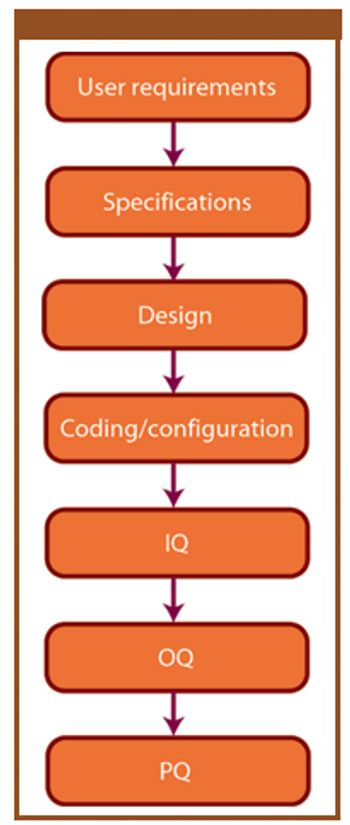
ePT--the Electronic Newsletter of Pharmaceutical Technology
The US Food and Drug Administration issued a rule that clarifies its requirements for current good manufacturing practices for aseptic processing, water standards, and verifications standards.