The presentation details the addition of triethylamine to the guideline on toxic solvents.
Regulatory Oversight and Compliance
Latest News
Advertisement
Advertisement
The agency published an overview of marketing authorizations made in 2016.
A new FDA Q&A document released on Jan. 12, 2016 describes 180-day exclusivity for generic-drug manufacturers and explains the number of conditions under which an abbreviated new drug applicant (ANDA) submitting a paragraph IV certification would forfeit eligibility to be the authorized generic manufacturer of a drug.
EDQM outlines the steps it will take to implement the ICH Q3D guideline on elemental impurities.
In a blog post, Robert Califf and Rita Nalubola discuss the agency’s approach to the use of genome-edited products.
An FDA guidance on biosimilar naming garnered mixed responses from the Biosimilars Forum, the American College of Rheumatology, and Pfenex.
FDA published a flurry of guidance documents in late 2016.
The agency plans on publishing more than 100 new or revised guidance documents in 2017.
The partnership will focus on providing practical information to clients on the development of biologics and vaccines.
The company was cited by FDA for violations of sterile processing GMPs.
FDA’s Center for Drug Evaluation and Research (CDER) approved 22 new molecular entities (NMEs) in 2016, according to a January 4, 2016 release from the agency. Of the 22 NMEs, 12 were large-molecule therapeutics. Of CDER’s 45 novel drug approvals in 2015, 17 were considered large-molecule therapeutics (larger than 900 Daltons).In 2016, 11 of the 12 large-molecule drugs that were approved were biologics. This includes seven monoclonal antibodies (mAbs), one hormone, and three DNA-derived medications. The remaining large-molecule medication was a diagnostic agent.

FDA plans to advance initiatives for ensuring reliable production of drugs and biologics in 2017.

A four-stage process to successfully make the switch from paper to electronic batch records is presented.
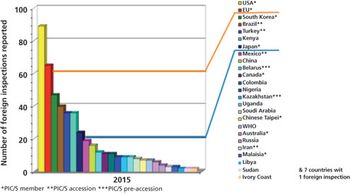
The European Federation of Pharmaceutical Industries and Associations (EFPIA) has conducted an annual survey of GMP/GDP inspections at sites and affiliates among its member companies from 2003 until the present time. This article describes findings of the EFPIA annual survey of site inspections among its member companies.
How involved should HTA bodies be in assessing cost effectiveness and reimbursements?
The agency has published its refuse-to-receive standards guidance for abbreviated new drug applications.
The agency put Baoying County Fukang Medical Appliance Co., Ltd. on import alert after observing violations at the company’s Yangzhou City facility.
The companies entered a manufacturing agreement for the future commercial production of Lenti-D and LentiGlobin product candidates.
The agency announces that 81 medicines overall were recommended in 2016.
FDA’s Center for Drug Evaluation and Research makes plans for implementation of the 21st Century Cures Act that include patient-focused drug development.
Congress enacted the 21st Century Cures legislation, which shores FDA operations and supports biomedical research at the National Institutes of Health.
FDA issued a warning letter to Interquim, SA for CGMP deviations at its Barcelona API facility.
Dara Corrigan examines the Mutual Reliance Initiative as a method for expanding FDA’s inspection capabilities in Europe and beyond.
Susan Schniepp, distinguished fellow at Regulatory Compliance Associates, discusses training personnel on a limited budget.

Airlocks, gowning rooms, and transition spaces have different uses and should be considered separately in cGMP pharmaceutical facility design.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Everything to Know about BMS & NVidia’s AI Supercomputer
2
Brent Wilhelm on Cold Chain Pharmaceuticals Reshaping Shipping Needs
3
Novo Nordisk Sues Eli Lilly Over Zepbound and Mounjaro Ads, Alleging Omitted GLP-1 Dosing Data
4
Can AI Fix Pharma's Last-Mile Delivery Blind Spot?
5