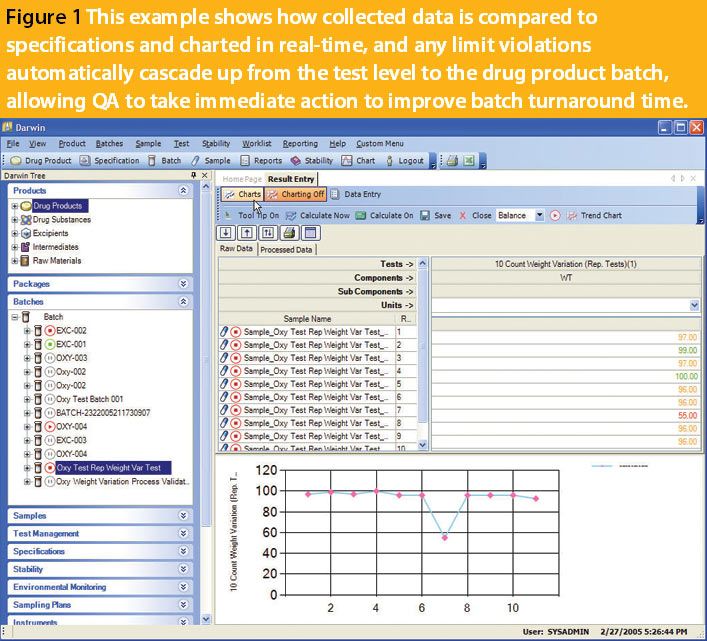

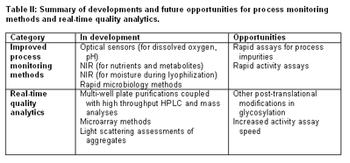
ePT--the Electronic Newsletter of Pharmaceutical Technology
"The better we understand the relationship between process parameters and product attributes, the better control we'll have over product quality," said Beth Fowler, PhD, during Tuesday?s session on process monitoring at the AAPS Annual Meeting.