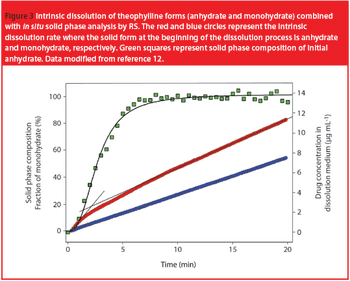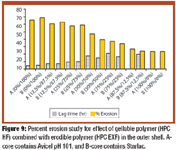

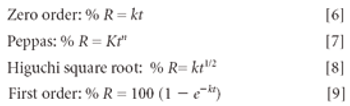
Hydrogels are biocompatible drug delivery systems by which the physical properties can be controlled by the cross-linking density. Hydrogels were prepared by copolymerization of acrylic acid monomers in the presence of poly(ethylene glycol)(PEG) to form polyethylene diacrylate (PEDGA). Various molecular weights of PEGs were used for the synthesis of PEGDA to study the effect of molecular weight of PEG on the properties of hydrogels. These hydrogels were further characterized for free water, swelling behavior, water diffusion, drug loading, and drug release profile. By analyzing the swelling behavior and release pattern of the hydrogels, the authors show that these systems can be suitably used for controlled delivery of drugs.