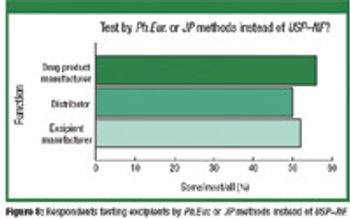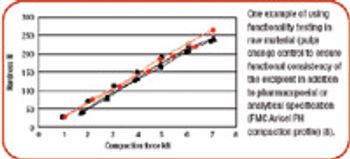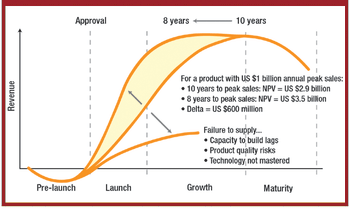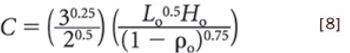


ePT--the Electronic Newsletter of Pharmaceutical Technology
Arlington, VA (Sept. 12)-At the American Association of Pharmaceutical Scientists meeting here, "e;Real World Applications of PAT and QbD in Drug Process Development and Approval" (Sept. 11-12), chemical engineer and process modeler Michael L. Thompson, PhD, described how Procter & Gamble (West Chester, OH, www.pg.com) applies these mathematical tools to increase product quality and reduce development and trouble-shooting time for consumer and pharmaceutical products.