
Under US regulations, such as the Medicare Access and CHP Reauthorization Act of 2015 (MACRA), part of any US physician’s reimbursement will be based on patient outcomes.
Quality Systems
Latest News
Advertisement
Advertisement

Value-based medicine is putting patients at the center of pharmaceutical R&D and forcing the industry to move from treatment to prevention.
The author discusses the impact of prefilled syringe product contact materials and the filling and stoppering process on protein aggregates.

Solid-phase extraction has several advantages over liquid/liquid extraction for extractables and leachables studies.
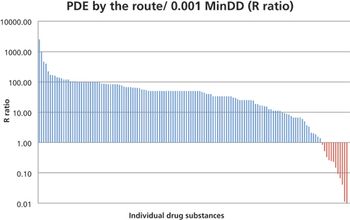
In this study, the authors investigated the relationship between the 0.001 MinDD and the PDE values for 140 drug substances as an attempt to identify high-risk groups of products for patient safety. This comparison can serve as a method for prioritization of APIs for development of PDEs.
The agency has issued more than 90 warning letters over the past 10 years to companies selling fraudulent cancer treatments.
On April 28, 2017, the European Medicines Agency announced that it met with members of the heads of the National Competent Authorities (NCAs) of the member states of the European Union to discuss the United Kingdom’s exit from the EU (Brexit) and how the agency and the member states will handle the evaluation and monitoring of drugs going forward.
Truxton, Inc. is voluntarily recalling one lot of Phenobarbital Tablets, USP, 15 mg because of a labeling error on declared strength.
FDA sent a warning letter to Teva Pharmaceutical and Chemical (Hangzhou) Co. Ltd. after an inspection found the company failed to monitor and control quality.
April 24–30, 2017 is European Immunization Week in Europe, and in a statement on the European Medicines Agency (EMA) website dated April 25, 2017, Executive Director Guido Rasi stressed the importance of vaccinations in preventing and controlling disease. Rasi acknowledged that fear created from incorrect information presented by unreliable sources has created a lack of trust in vaccines. But he highlighted that Europe monitors and records information on the safety of medicines, including vaccines.
The company is recalling one lot of 25% Dextrose injection after particulate matter was found within an internal sample syringe.
Sandoz, a Novartis division, announced that the Committee for Medicinal Products for Human Use (CHMP) has adopted positive opinions recommending the approval of its biosimilars rituximab and etanercept in Europe, for the same indications as the respective reference medicines.
The agency approved Renflexis, a biosimilar to Janssen’s blockbuster rheumatoid arthritis treatment.
The agency is warning about the potential threat of respiratory depression in children who take medicines with codeine or tramadol.
Equipment and Processing Report
In implementing quality by design for drug formulation, it is crucial to identify the critical properties of excipients and understand how their variation affects the final drug product.
PTSM: Pharmaceutical Technology Sourcing and Management
Pharmaceutical Technology spoke with Brad Pedrow and Rajesh Singh of Deloitte Consulting to discuss serialization implementation, and what to expect as the DSCSA deadline approaches.
igital tracking of overall equipment effectiveness can improve efficiency.
FDA denies the NDA for the RA drug developed by Eli Lilly and Incyte, citing the need for more data.
Standard Homeopathic Company is recalling all lots of Hyland’s Baby Teething Tablets and Hyland’s Nighttime Baby Teething Tablets due to inconsistent amounts of belladonna alkaloids.
Integrating EBRs with MES reduces time to market, cuts costs, and enhances compliance performance.
Pharmaceutical Technology spoke with CPhI North America presenter Jonathan Helfgott to discuss navigating GDUFA and helpful tips for submitting successful ANDAs.
At CPhI North America 2017, the US Pharmacopeial Convention will be discussing its upcoming revision and modernization of the standards for elemental and organic impurities.
The agency cited the company’s India facility for batch failures and data integrity problems.
The new guide offers guidance on how to ensure data and records are complete, consistent, secure, accurate, and available throughout their lifecycle.
Pharmaceutical Technology spoke with CPhI North America presenters Ben Locwin, PhD, MBA, MBB, president at Healthcare Science Advisors, and Tom Fox, principal at Advanced Compliance Solutions, to discuss drug pricing, compliance challenges, and corporate social responsibility in the bio/pharmaceutical industry.
Advertisement
Advertisement
Trending on Pharmaceutical Technology
1
Novo Nordisk Sues Eli Lilly Over Zepbound and Mounjaro Ads, Alleging Omitted GLP-1 Dosing Data
2
Can AI Fix Pharma's Last-Mile Delivery Blind Spot?
3
Everything to Know about BMS & NVidia’s AI Supercomputer
4
From Predictions to Reality: Mike Stenberg Assesses Pharma Manufacturing's First Half of 2026
5