



Connect with pharmaceutical and healthcare regulatory authorities around the world via this directory.
Connect with pharmaceutical and healthcare regulatory authorities around the world via this directory.

Find links to pertinent regulatory and standard setting resources, guidance documents, and guidelines.

Long a staple for quality by design and solid dosage form manufacturing, design of experiments is becoming an integral part of biopharma upstream process development.

Warning letters tell the tale of missteps by drug companies and offer a path to compliance for quality teams that monitor these enforcement actions.

Too narrow a focus on regulatory compliance may prevent organizations from embracing-and profiting from-quality and operational excellence.

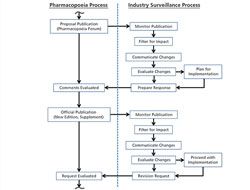
An effective surveillance program for monitoring the activities of pharmacopoeias around the world requires processes, people, and tools from across a company.
An effective surveillance program for monitoring the activities of pharmacopoeias around the world requires processes, people, and tools from across a company.
This article describes the revision process and the resulting publication of proposed and official updates for pharmacopoeias around the world.
According to a consent decree of permanent injunction issued in September 2019, the health and wellness companies must recall and stop distributing products until the companies comply with the Federal Food, Drug, and Cosmetic Act and other requirements included in the consent decree.
While labor and tariff reforms in the revised North American trade agreement may have more visible impacts on the United States economy, the final document levels a major blow to exclusivity and patent protections important to innovator biotech and pharma companies.
FDA has an official new leader, following a Senate vote to confirm Stephen Hahn as the agency’s next commissioner.
The biologic is a novel bone builder that has a dual effect of increasing bone formation and reducing bone loss.
The agency has approved three applications for generic versions of Gilenya (fingolimod), Novartis’ blockbuster multiple sclerosis drug.
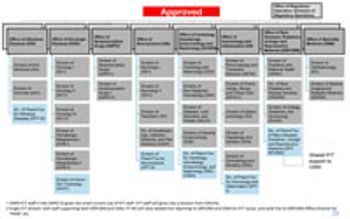
FDA’s Office of New Drugs restricting aims to improve scientific exchange and information sharing among review professionals.
A NASEM report stresses the importance of information sharing by biopharma companies and cooperation among regulatory authorities.

Applying lessons of raw materials’ characterization and supply-chain control from the semiconductor industry allow more rigorous control of the biopharmaceutical manufacturing process.
Regulators are facing huge challenges on how to deal with the digitalization transformation occurring in the healthcare and pharmaceutical sectors.

Communications and planning are crucial to recovering from supply, operations, and facility disruptions.

As equipment evolves and use of PAT increases, DOE is becoming an integral part of upstream bioprocess development.
Problems in assuring reliable drug quality and supply dampens progress in bringing lifesaving therapies to market.
Investigating deviations of combination products needs to accommodate both the drug and device components, says Susan J. Schniepp, executive vice-president of post-approval pharma and distinguished fellow, Regulatory Compliance Associates.

This article discusses reduced sampling and testing of starting materials or components. Different strategies are presented to reduce the workload at the steps from sampling to release. Viewpoints from the different pharmacopoeias and regulatory authorities, as well as selected literature, are reviewed.
The agency sent warning letters to 15 companies for illegally selling cannabidiol products.
The agency sent a warning letter to Torrent Pharma after an inspection found violations of current good manufacturing practices that included a failure to thoroughly investigate batch failures.
The guidance provides recommendations for the development and quality information that should be included in NDA and ANDA applications.