



ePT--the Electronic Newsletter of Pharmaceutical Technology
GSK Gains Approval for Rotavirus Vaccine in Infants; FDA Review Extended for Merck’s Shingles Vaccine
The evolution of therapeutic modalities drives the adoption of single-use technologies.

Despite its understandable hesitancy, the pharma industry is facing a need for more widespread adoption of cloud-based solutions.

Given the criticality of fill/finish processes, it is clear that automation is the next technological step.

ePT--the Electronic Newsletter of Pharmaceutical Technology
GSK Gains Approval for Rotavirus Vaccine in Infants; FDA Review Extended for Merck’s Shingles Vaccine

Pharmaceutical Technology Europe
The PAT guidance indicates a variety of risk-based approaches to managing the introduction of on-line analysers into existing processes with the aim of minimizing the regulatory burden for the manufacturer and encouraging innovation.

Pharmaceutical Technology Europe
Clean rooms are areas in which it is essential that microorganisms are not allowed to proliferate because they could contaminate pharmaceuticals and directly affect human health.

Pharmaceutical Technology Europe
Pharma companies are forming alliances with less established biotech companies, which significantly drive the technological innovation and the overall market growth.
ePT--the Electronic Newsletter of Pharmaceutical Technology
FDA Recommends Changes for 2006-2007 Flu Vaccine
ePT--the Electronic Newsletter of Pharmaceutical Technology
Baxter Wins Contract to Develop Cell-Based H5N1 Vaccine

Applikon Biotechnology
ePT--the Electronic Newsletter of Pharmaceutical Technology
Cambrex (East Rutherford, NJ) to produce Geron (Menlo Park, CA) telomerase anti-cancer vaccine. Generex (Toronto, ON.) files IND for synthetic vaccine to stimulate cell-mediated immunity to avian influenza. Dow Agrosciences (Indianapolis, IN) wins USDA approval for veterinary vaccine, the first manufactured via plant cell culture. G-8 nations pledge billons for vaccine production.
ePT--the Electronic Newsletter of Pharmaceutical Technology
The Pharmaceutical Industry has been slow in adopting radio frequency technology (RFID) to help control diversion and counterfeiting, according to a recent study by ABI Research (Oyster Bay, NY, www.abiresearch.com). In fact, only 10 drug products are expected to be shipped with RFID tags or smart chips embedded in the labeling in the coming year.
ePT--the Electronic Newsletter of Pharmaceutical Technology
Merck wins approval for rotavirus vaccine as Sanofi ships investigational H5N1 vaccine to NIH and CDC explores new flue diagnostics and novel vaccine-production technology.
ePT--the Electronic Newsletter of Pharmaceutical Technology
Dow AgroSciences Receives Regulatory Approval for Plant-Made Vaccine
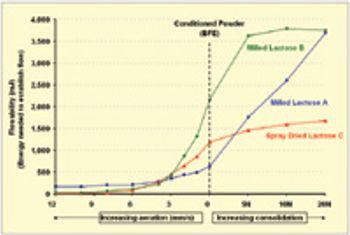
The pharmaceutical industry's focus on process understanding, monitoring, and control is driving manufacturers to take greater steps toward identifying possible manufacturing bottlenecks earlier in the development process. For tablet, capsule, and excipient producers, such efforts include taking a closer look at the flow-ability of their powders.

Enterprise information integration delivers needed real-time capability to operational business intelligence.

FDA is proposing revisions to the drug user fee program while it weighs changes for drug safety initiatives and expanded vaccine production.
ePT--the Electronic Newsletter of Pharmaceutical Technology
Cell-Grown Vaccine Protects Against Avian Flu Virus
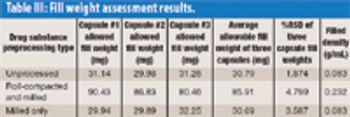
Using a novel automated microfilling system, the authors demonstrate that roller compaction followed by milling is a viable preprocessing technique for high-dose chemical-in-capsule dosage forms. The process results in higher bulk and tapped densities for drug substances compared with milling alone.

Pharmaceutical Technology Europe
The regulating authorities have, it seems, a preference for the application of multimembrane combinations to maximize organism retentions.

Pharmaceutical Technology Europe
There are over 250 operations in the EU in various stages of development involving tissue engineering, regeneration and subsequent attempts at commercialization.
Pharmaceutical Technology Europe
The type of robot used for placing and stacking the BFS cards is important. Conventional multi-axes designs have limited flexibility, often combined with high inertia that limits operating speeds.

Though dissolution testing has been under scrutiny, it is still a powerful test method.
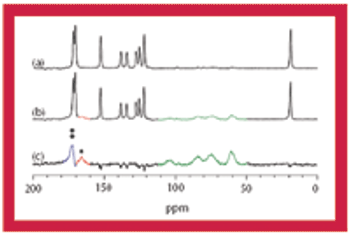
This article discusses the advantages and disadvantages of using solid-state NMR spectroscopy for the analysis of pharmaceutical solids.
ePT--the Electronic Newsletter of Pharmaceutical Technology
Pfizer Combats Counterfeiters with RFID
ePT--the Electronic Newsletter of Pharmaceutical Technology
Sanofi Pasteur and BD Team Up to Produce Microneedle Vaccines

Pharmaceutical Technology Europe
There is a rapidly growing ageing population that requires sophisticated medical devices and newer drugs. This is likely to result in an increase in the use of robotics to improve manufacturing efficiency. This article looks at the role of SCARA robots in pharmaceutical plants and laboratories.
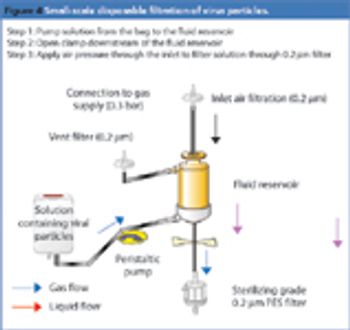
Pharmaceutical Technology Europe
The medical industry was the first to understand the benefits of using disposable devices, such as needles and syringes, to prevent risks of cross contamination. The technology was then extended to blood transfusion activities, and it was only 10–15 years ago that the biopharmaceutical industry started to use disposables. Initially, most of the applications were limited to storage, involving bags, tubing and filter capsules. Since then, significant progress has been made in the polymer and plastics industry; in particular, a number of organic polymers have been developed that are resistant to gamma irradiation, autoclaving and even sterilization-in-place, rendering the technology attractive and usable by the biopharmaceutical industry. Now, the industry is moving beyond storage-focused disposable technologies to more complex processing applications.